 Dr. Tuuli Lappalainen is a postdoctoral researcher at Stanford University, where she works on functional genetic variation in human populations and specializes in population-scale RNA-sequencing. She kindly agreed to write a guest post on her recent publication in Nature, “Uncovering functional variation in humans by genome and transcriptome sequencing”, which describes work done while she was at the University of Geneva. -DM
Dr. Tuuli Lappalainen is a postdoctoral researcher at Stanford University, where she works on functional genetic variation in human populations and specializes in population-scale RNA-sequencing. She kindly agreed to write a guest post on her recent publication in Nature, “Uncovering functional variation in humans by genome and transcriptome sequencing”, which describes work done while she was at the University of Geneva. -DM
In a paper published online today in Nature we describe results of the largest RNA-sequencing study of multiple human populations to date, and provide a comprehensive map of how genetic variation affects the transcriptome. This was achieved by RNA-sequencing of individuals that are part of the 1000 Genomes sample set, thus adding a functional dimension to the most important catalogue of human genomes. In this blog post, I will discuss how our findings shed light on genetic associations to disease.
As genome-wide studies are providing an increasingly comprehensive catalog of genetic variants that predispose to various diseases, we are faced with a huge challenge: what do these variants actually do in the cell? Understanding the biological mechanisms underlying diseases is essential to develop interventions, but traditional molecular biology follow-up is not really feasible for the thousands of discovered GWAS loci. Thus, we need high-throughput approaches for measuring genetic effects at the cellular level, which is an intermediate between the genome and the disease. The cellular trait most amenable for such analysis is the transcriptome, which we can now measure reliably and robustly by RNA-sequencing (as shown by our companion paper in Nature Biotechnology).
In this project, several European institutes of the Geuvadis Consortium sequenced mRNA and small RNA from lymphoblast cell lines from 465 individuals that are in the 1000 Genomes sample set. The idea of gene expression analysis of genetic reference samples is not new (see e.g. papers by Stranger et al., Pickrell et al. and Montgomery et al.), but the bigger scale and better quality enables discovery of exciting new biology.
Regulatory variants underlying GWAS signals
Our first striking observation was that over one half of measured genes are affected by common genetic variation in human populations – called expression quantitative trait loci or eQTLs. Regulatory associations are not like GWAS studies where you are lucky to find a handful of significant hits; regulatory variation is literally (almost) everywhere – it’s the rule, not the exception.
The vast majority these regulatory variants won’t have any effect on the phenotype at the individual level, but some of them do. The first obvious question was how many known GWAS variants are eQTLs in our study, and indeed pretty many of them are – 16%. So does this prove that in all these GWAS regions we have identified the regulatory change as the cellular mechanism that drives the disease? Unfortunately the answer is no. Regulatory associations are so common that the expected overlap just by chance is as high as 11%. This means that your favorite GWAS variant having a significant regulatory association is very far from sufficient proof of it being the biological mechanism of the disease or trait. The same applies to overlap with for example ENCODE annotations, by the way. This is not overcautious small print. We’ve basically reversed the problem of having hardly any clue of functional mechanisms to having too many putative functions. We’ve found the haystack.
How can we solve this problem? Luckily, there are statistical methods to analyze the two association signals in the same genomic region to find out if the gene expression association is likely to be causal to the disease association. You still can’t be 100% sure, but that is much smaller print. And we do find an enrichment of such a signal, as in previous studies – telling us that regulatory changes are enriched for being causal biological mechanisms underlying GWAS signals.
From associated regions to causal variants
We can take this analysis an important step further to pinpoint likely causal variants. Thus far, nearly all association studies have used data from SNP arrays that measure only a subset of all the common variants. This works fine for identifying more or less broad regions of the genome that have a variant somewhere that changes the function of the genome such that it predisposes to the trait in question. However, usually there’s no clue what the precise causal variant is and what its exact properties are.
The first step in finding the causal variants is getting genome sequencing data, which is what we have in our study. We show that we have pretty good power to pinpoint causal regulatory variants in many of the loci, which is great news for understanding mechanisms of genome regulation. This has a cool application for dozens of GWAS loci that are driven by a regulatory association: by discovering the putative causal regulatory variant from our association data, we’re at the same time pinpointing the likely causal GWAS variant as well. Thus, combining genome sequencing and cellular phenotype data can give us information not only of the biological mechanisms underlying GWAS associations, but also identify the likely causal variants.
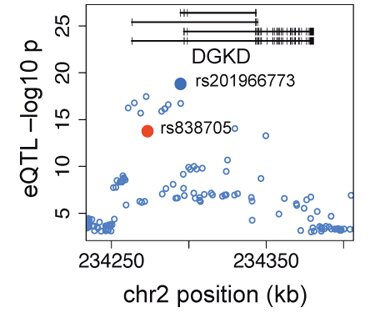 The example of the DGKD gene (left) illustrates the power of our eQTL data to map causal functional variants. The plots show the eQTL association landscape, where the top eQTL variant rs201966773 is the most likely causal variant. This variant is a 2bp insertion that is not genotyped by any SNP arrays, and overlaps several regulatory elements close to the transcript start site of some of the transcripts of the gene. The rs838705 variant marked in red is a GWAS variant associated to calcium levels – and the eQTL analysis suggests that rs201966773 is the most likely causal variant for this association signal.
The example of the DGKD gene (left) illustrates the power of our eQTL data to map causal functional variants. The plots show the eQTL association landscape, where the top eQTL variant rs201966773 is the most likely causal variant. This variant is a 2bp insertion that is not genotyped by any SNP arrays, and overlaps several regulatory elements close to the transcript start site of some of the transcripts of the gene. The rs838705 variant marked in red is a GWAS variant associated to calcium levels – and the eQTL analysis suggests that rs201966773 is the most likely causal variant for this association signal.
Where to next?
In this study we have integrated genome and transcriptome sequencing data to understand the landscape of functional variation in human populations. In addition to our scientific discoveries, this is an extremely valuable open-access data set for the human genetics community, as it links directly to the 1000 Genomes data that is used by nearly all human genetics projects. Since our pre-publication data release in November 2012, the data set has already been downloaded thousands of times, and we’ve put a lot of effort into open data sharing by having a browser and even opening our project wiki for the public.
This paper is a big step forward, but we’re still far from full understanding of how genetic variation affects the transcriptome and how this affects human disease. One important challenge is cellular effects of rare and loss-of-function variants, which we address only briefly in this paper. Furthermore, other projects such as GTEx are describing transcriptome variation and its genetic causes in large variety of human tissues. These studies will provide very exciting insights into genome function in the not-too-distant future, and I’m sure that these results will be discussed on this blog as well.
This study and other projects analyzing cellular phenotypes in the general human population are providing the baseline of the general population spectrum of functional genetic variation and transcriptome variation, which is essential to be able to distinguish the cases where things go wrong and cause disease. At the same time as we move forward with basic research, it is important to push for clinical applications to target cellular perturbations leading to disease, and develop approaches for personalized transcriptomics to better interpret personalized genomes.








 RSS
RSS Twitter
Twitter
Briefly, given recent discoveries in substantial aneuploidy in human tissue, particularly human neurons, is there an issue of the applicability of transcriptome measurements from lymphoblasts as applied to other tissues that vary both in aneuploidy (from cell to cell in neural tissues) as well as elsewhere? Via recent discussions, it appears some scientists are suggesting that substantial brain tissue aneuploidy may be a significant complicating factor in genetic and transcriptome measurements.
http://www.academia.edu/824808/Failed_clearance_of_aneuploid_embryonic_neural_progenitor_cells_leads_to_excess_aneuploidy_in_the_Atm-deficient_but_not_the_Trp53-deficient_adult_cerebral_cortex
http://www.jneurosci.org/content/25/9/2176.full.pdf
Brief follow-up:
Measurement of aneuploidy in the genome is not my area but my understanding (per recent personal communication) is that much more sophisticated measurements of aneuploidy are becoming feasible that may clarify whether my point above is significant of not.
Apparently a group has already done a paper on the issue I asked about:
http://www.ncbi.nlm.nih.gov/pubmed/22968442
Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells.
Max Planck Institute of Biochemistry